
Page 41 - Haematologica April 2020
P. 41
RUNX1 variant curation
A
B
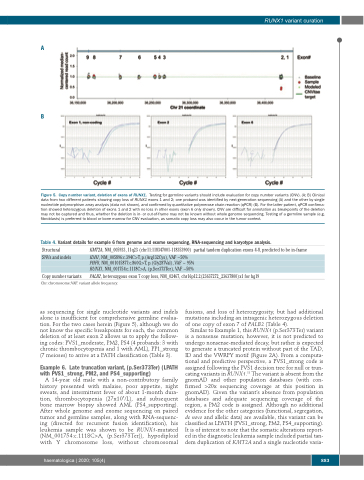
Figure 5. Copy number variant, deletion of exons of RUNX1. Testing for germline variants should include evaluation for copy number variants (CNV). (A; B) Clinical data from two different patients showing copy loss of RUNX1 exons 1 and 2; one proband was identified by next-generation sequencing (A) and the other by single nucleotide polymorphism array analysis (data not shown), and confirmed by quantitative polymerase chain reaction (qPCR) (B). For the latter patient, qPCR confirma- tion showed heterozygous deletion of exons 1 and 2 with no loss in other exons (exon 6 only shown). CNV are difficult for annotation as breakpoints of the deletion may not be captured and thus, whether the deletion is in- or out-of-frame may not be known without whole genome sequencing. Testing of a germline sample (e.g. fibroblasts) is preferred to blood or bone marrow for CNV evaluation, as somatic copy loss may also occur in the tumor context.
Table 4. Variant details for example 6 from genome and exome sequencing, RNA-sequencing and karyotype analysis.
Structural
SNVs and indels
as sequencing for single nucleotide variants and indels alone is insufficient for comprehensive germline evalua- tion. For the two cases herein (Figure 5), although we do not know the specific breakpoints for each, the common deletion of at least exon 2 allows us to apply the follow- ing codes: PVS1_moderate, PM2, PS4 (4 probands: 3 with chronic thrombocytopenia and 1 with AML), PP1_strong (7 meioses) to arrive at a PATH classification (Table 3).
Example 6. Late truncation variant, (p.Ser373Ter) (LPATH with PVS1_strong, PM2, and PS4_supporting)
A 14-year old male with a non-contributory family history presented with malaise, poor appetite, night sweats, and intermittent fever of about 1-month dura- tion, thrombocytopenia (27x109/L), and subsequent bone marrow biopsy showed AML (PS4_supporting). After whole genome and exome sequencing on paired tumor and germline samples, along with RNA-sequenc- ing (directed for recurrent fusion identification), his leukemia sample was shown to be RUNX1-mutated (NM_001754:c.1118C>A, (p.Ser373Ter)), hypodiploid with Y chromosome loss, without chromosomal
KMT2A, NM_005933, 11q23 (chr11:118347001-118353900) partial tandem duplication exons 4-8, predicted to be in-frame
IDH1, NM_005896:c.394C>T, p.(Arg132Cys), VAF ~50% PHF6, NM_001015877:c.860G>T, p.(Gly287Val), VAF ~ 95% RUNX1, NM_001754:c.1118C>A, (p.Ser373Ter), VAF ~50%
PALB2, heterozygous exon 7 copy loss, NM_02467, chr16p12.2(23637272_23637800)x1 for hg19 Chr: chromosome;VAF: variant allele frequency.
Copy number variants
fusions, and loss of heterozygosity, but had additional mutations including an intragenic heterozygous deletion of one copy of exon 7 of PALB2 (Table 4).
Similar to Example 1, this RUNX1 (p.Ser373Ter) variant is a nonsense mutation; however, it is not predicted to undergo nonsense-mediated decay, but rather is expected to generate a truncated protein without part of the TAD, ID and the VWRPY motif (Figure 2A). From a computa- tional and predictive perspective, a PVS1_strong code is assigned following the PVS1 decision tree for null or trun- cating variants in RUNX1.18 The variant is absent from the gnomAD and other population databases (with con- firmed >20x sequencing coverage at this position in gnomAD). Given the variant’s absence from population databases and adequate sequencing coverage of the region, a PM2 code is assigned. Although no additional evidence for the other categories (functional, segregation, de novo and allelic data) are available, this variant can be classified as LPATH (PVS1_strong, PM2, PS4_supporting). It is of interest to note that the somatic alterations report- ed in the diagnostic leukemia sample included partial tan- dem duplication of KMT2A and a single nucleotide varia-
haematologica | 2020; 105(4)
883
