
Page 280 - Haematologica April 2020
P. 280
V. Daidone et al.
A
B
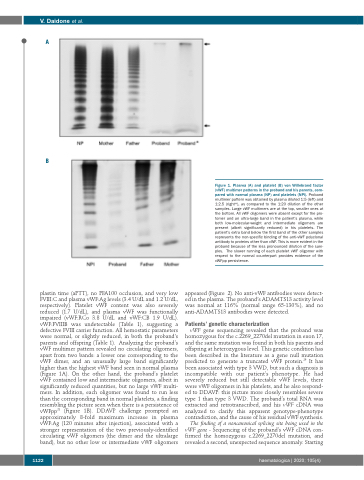
plastin time (aPTT), no PFA100 occlusion, and very low FVIII:C and plasma vWF:Ag levels (3.4 U/dL and 1.2 U/dL, respectively). Platelet vWF content was also severely reduced (1.7 U/dL), and plasma vWF was functionally impaired (vWF:RCo 3.8 U/dL and vWF:CB 1.9 U/dL). vWF:FVIIIB was undetectable (Table 1), suggesting a defective FVIII carrier function. All hemostatic parameters were normal, or slightly reduced, in both the proband’s parents and offspring (Table 1). Analyzing the proband’s vWF multimer pattern revealed no circulating oligomers, apart from two bands: a lower one corresponding to the vWF dimer, and an unusually large band significantly higher than the highest vWF band seen in normal plasma (Figure 1A). On the other hand, the proband’s platelet vWF contained low and intermediate oligomers, albeit in significantly reduced quantities, but no large vWF multi- mers. In addition, each oligomer was found to run less than the corresponding band in normal platelets, a finding resembling the picture seen when there is a persistence of vWFpp28 (Figure 1B). DDAVP challenge prompted an approximately 8-fold maximum increase in plasma vWF:Ag (120 minutes after injection), associated with a stronger representation of the two previously-identified circulating vWF oligomers (the dimer and the ultralarge band), but no other low or intermediate vWF oligomers
appeared (Figure 2). No anti-vWF antibodies were detect- ed in the plasma. The proband’s ADAMTS13 activity level was normal at 116% (normal range 65-130%), and no anti-ADAMTS13 antibodies were detected.
Patients’ genetic characterization
vWF gene sequencing revealed that the proband was homozygous for the c.2269_2270del mutation in exon 17, and the same mutation was found in both his parents and offspring at heterozygous level. This genetic condition has been described in the literature as a gene null mutation predicted to generate a truncated vWF protein.29 It has been associated with type 3 VWD, but such a diagnosis is incompatible with our patient’s phenotype. He had severely reduced but still detectable vWF levels, there were vWF oligomers in his platelets, and he also respond- ed to DDAVP: this picture more closely resembles severe type 1 than type 3 VWD. The proband’s total RNA was extracted and retrotranscribed, and his vWF cDNA was analyzed to clarify this apparent genotype-phenotype contradiction, and the cause of his residual vWF synthesis.
The finding of a noncanonical splicing site being used in the vWF gene - Sequencing of the proband’s vWF cDNA con- firmed the homozygous c.2269_2270del mutation, and revealed a second, unexpected sequence anomaly. Starting
Figure 1. Plasma (A) and platelet (B) von Willebrand factor (vWF) multimer patterns in the proband and his parents, com- pared with normal plasma (NP) and platelets (NPl). Proband multimer pattern was obtained by plasma diluted 1:5 (left) and 1:2.5 (right*), as compared to the 1:20 dilution of the other samples. Large vWF multimers are at the top, smaller ones at the bottom. All vWF oligomers were absent except for the pro- tomer and an ultra-large band in the patient’s plasma, while both low-molecular-weight and intermediate oligomers are present (albeit significantly reduced) in his platelets. The patient’s extra band below the first band of the other samples represents the non-specific binding of the anti-vWF polyclonal antibody to proteins other than vWF. This is more evident in the proband because of the less pronounced dilution of the sam- ples. The slower running of each platelet vWF oligomer with respect to the normal counterpart provides evidence of the vWFpp persistence.
1122
haematologica | 2020; 105(4)
