
Page 262 - Haematologica April 2020
P. 262
S.I. Obydennyi et al.
A
B
D
E
C
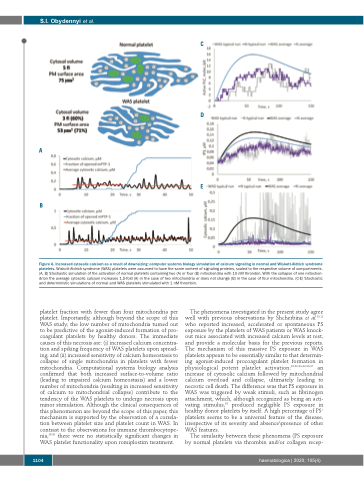
Figure 6. Increased cytosolic calcium as a result of downsizing: computer systems biology simulation of calcium signaling in normal and Wiskott-Aldrich syndrome platelets. Wiskott-Aldrich syndrome (WAS) platelets were assumed to have the same content of signaling proteins, scaled to the respective volume of compartments. (A, B) Stochastic simulation of the activation of normal platelets containing two (A) or four (B) mitochondria with 10 nM thrombin. With the collapse of one mitochon- drion the average cytosolic calcium increases 1.5-fold (A) in the case of two mitochondria or does not change (B) in the case of four mitochondria. (C-E) Stochastic and deterministic simulations of normal and WAS platelets stimulated with 1 nM thrombin.
platelet fraction with fewer than four mitochondria per platelet. Importantly, although beyond the scope of this WAS study, the low number of mitochondria turned out to be predictive of the agonist-induced formation of pro- coagulant platelets by healthy donors. The immediate causes of this necrosis are: (i) increased calcium concentra- tion and spiking frequency of WAS platelets upon spread- ing; and (ii) increased sensitivity of calcium homeostasis to collapse of single mitochondria in platelets with fewer mitochondria. Computational systems biology analysis confirmed that both increased surface-to-volume ratio (leading to impaired calcium homeostasis) and a lower number of mitochondria (resulting in increased sensitivity of calcium to mitochondrial collapse) contribute to the tendency of the WAS platelets to undergo necrosis upon minor stimulation. Although the clinical consequences of this phenomenon are beyond the scope of this paper, this mechanism is supported by the observation of a correla- tion between platelet size and platelet count in WAS. In contrast to the observations for immune thrombocytope- nia,29,35 there were no statistically significant changes in WAS platelet functionality upon romiplostim treatment.
The phenomena investigated in the present study agree well with previous observations by Shcherbina et al.10,11 who reported increased, accelerated or spontaneous PS exposure by the platelets of WAS patients or WAS knock- out mice associated with increased calcium levels at rest, and provide a molecular basis for the previous reports. The mechanism of this massive PS exposure in WAS platelets appears to be essentially similar to that determin- ing agonist-induced procoagulant platelet formation in physiological potent platelet activation:18,26,33,34,36,37 an increase of cytosolic calcium followed by mitochondrial calcium overload and collapse, ultimately leading to necrotic cell death. The difference was that PS exposure in WAS was triggered by weak stimuli, such as fibrinogen attachment, which, although recognized as being an acti- vating stimulus,38 produced negligible PS exposure in healthy donor platelets by itself. A high percentage of PS+ platelets seems to be a universal feature of the disease, irrespective of its severity and absence\presence of other WAS features.
The similarity between these phenomena (PS exposure by normal platelets via thrombin and/or collagen recep-
1104
haematologica | 2020; 105(4)
