
Page 93 - 2019_12-Haematologica-web
P. 93
Somatic variants in CML-CP as predictive biomarker
apy, we selected a cohort enriched for non-responders (approx. 45% in each of the imatinib and 2G-TKI cohorts), which explains why the outcomes of patients in this study are inferior to those seen in unselected subjects.3,4,14 We believed our strategy of deliberately choosing equal pro- portions of responders and non-responders would maxi- mize our chances of detecting a biomarker, if such existed. Moreover, the use of CD34+ progenitor cells from patients at diagnosis, the population in which the clone capable of progression resides, would maximize our chance of yield- ing meaningful results and reduce heterogeneity. We are aware that, for clinical utilization, our data should be val- idated in whole-blood samples and in a larger cohort of newly-diagnosed individuals with CML.
Progression to blast crisis in CML is often attributed to underlying ‘genetic instability’, in part because this is increased in stem/progenitor cells from individuals with CML (especially in blast phase) compared to normal indi- viduals. BCRABL1-induced genomic aberrations and/or BCRABL1-independent pre-existing genetic lesions may then function as “amplifiers” of a genetically unstable phe- notype and thereby predispose to blastic transformation.45 However, our results, and those of others, suggest that the ‘mutator’ phenotype of CML is moderate compared with other cancers, particularly in chronic phase. We included genes in which somatic variants have been identified by WES21-24 including ASXL1, RUNX1, IKZF1, KDM2D, BCOR, IDH1/2, PHF6, TET2, KDM1A, KAT6A, SETBP1, SETD2 and found somatic variants in approximately 30% of newly-diagnosed individuals with CML-CP, similar to previous reports.25,27,28 We identified overlap with other studies of 14-30% but feel this can be explained, at least in part, by our focus on epigenetic regulators that resulted in the omission of some genes that have frequently been
found mutated in CML, such as TP53, and also by the readily available technology of targeted NGS at the start of this project.18,27,28 Concordance with other studies regarding specific variants was also limited, while 15 somatic variants in our study were COSMIC, mostly iden- tified in other hematologic neoplasms. Because most of the variants we identified affect epigenetic modifiers and genome-wide DNA methylation changes are reported in CML,32,46 a better understanding of the role of such epige- netic alteration should be complemented by genome-wide landscape of histone marks.
In this study, we report for the first time a correlation between somatic variants and survival of individuals with CML-CP. Others have reported the presence of somatic variants but have so far been unable to directly associate these with clinical outcome21-24,28 or limited the assessment to achievement of major molecular remission.25 One study found that a subset of variants (16 of 73) affecting epige- netic modifiers had an adverse impact on cytogenetic/molecular responses.27 Because of our use of extreme responders, and because of the availability of pro- longed follow up, our cohort contains patients who had experienced disease progression, with 20 patients devel- oping blast crisis over the period of observation. Eleven of these (8 of 13 and 3 of 7 on imatinib and 2G-TKI, respec- tively) had somatic variants. Absence of variants in the remaining subjects does not exclude their presence in genes absent from our panel, who could have structural variants/copy number variations such as IKZF1 deletions that would be identified by WES/whole-genome sequenc- ing whole-genome sequencing (WGS).
Although the two patient cohorts (treated with imatinib or 2G-TKI) were similar in their clinical characteristics at diagnosis and in the proportion of responders and non-
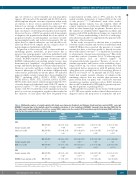
Table 3. Multivariate analysis of somatic variants with Sokal score, European Treatment and Outcome Study long-term survival (ELTS) score and BCRABL1 transcript type in the imatinib cohort for cumulative incidence of 3-log reduction in BCRABL1 transcripts from baseline (MR3) (by the Fine-Gray model) and probabilities of event-free survival (EFS), progression-free survival (PFS) and chronic myeloid leukemia (CML)-related sur- vival (by the Cox proportional hazard regression model).
MVA
Somatic variants at Dx Sokal score at Dx
MVA
Somatic variants at Dx ELTS score at Dx
MVA
Somatic variants at Dx
BCRABL1 transcript type at Dx
HR (95% CI)
P
HR (95% CI)
P
HR (95% CI)
P
HR (95% CI)
P
HR (95% CI)
P
HR (95% CI)
P
MR3 cumulative incidence
0.43 (0.19, 1.02) 0.054#
0.71 (0.48, 1.04) 0.080#
0.48 (0.20, 1.15) 0.098#
0.53 (0.30, 0.93) 0.028*
0.43 (0.19, 0.99) 0.049 * 0.90 (0.58, 1.39)
0.63
EFS probability
2.92 (1.32, 6.49) 0.008 ** 1.45 (0.88, 2.35) 0.14
2.53 (1.12, 5.71) 0.026 * 1.77 (1.08, 2.90) 0.025 *
3.21 (1.43, 7.19) 0.005 ** 1.15 (0.65, 2.03) 0.64
PFS probability
3.15 (1.06, 9.40) 0.040 * 2.31 (1.08, 4.95) 0.032 *
2.57 (0.85, 7.81) 0.095 .
2.10 (1.05, 4.22) 0.037 *
3.23 (1.87, 9.67) 0.036 * 0.97 (0.45, 2.11) 0.95
CML-related survival probability
3.09 (1.01, 9.69) 0.049 * 2.90 (1.27, 6.42) 0.011 *
2.60 (0.82, 8.25) 0.11
2.73 (1.31, 5.67) 0.007 **
3.30 (1.03, 10.5) 0.044 *
1.2 (0.53, 2.88) 0.62
†CI: confidence intervals; Dx: diagnosis; EFS: event-free survival; ELTS: EUTOS long-term survival; HR: hazard ratio; MR3: 3-log reduction in BCRABL1 transcripts from baseline; MVA: multivariate analysis; PFS: progression-free survival. #P<0.1; *P<0.05; **P<0.01.
haematologica | 2019; 104(12)
2407
